
Timisoara_Med 2022, 2022(1), 3; doi:10.35995/tmj20220103
Article
The Effects of Nitric Oxide Synthase Inhibitors in Acetaminophen-Induced Hepatic Injury in Mice
1
Department of Pharmacology, Faculty of Medicine, Muğla Sıtkı Koçman University, 48000 Mugla, Turkey; edipgcekic@mu.edu.tr
2
Department of Pathology, Faculty of Medicine, Muğla Sıtkı Koçman University, 48000 Mugla, Turkey; ozgurilhancelik@mu.edu.tr
3
Department of Biochemistry, Faculty of Medicine, Isparta Süleyman Demirel University, 32260 Isparta, Turkey; fbsirin@gmail.com
4
Department of Biochemistry, Faculty of Medicine, Muğla Sıtkı Koçman University, 48000 Mugla, Turkey; nigaryilmaz@mu.edu.tr
5
Department of Pharmacology, Faculty of Medicine, Hacettepe University, 06800 Ankara, Turkey; gsoydan@hacettepe.edu.tr.
*
Correspondence: edipgcekic@mu.edu.tr; Tel.: +90-252-211-4844
How to cite: Çekiç, E.G.; Filiz Başaran, N.; İlhan Çelik, O.; Şirin, F.B.; Yilmaz, N.; Soydan G. The Effects of Nitric Oxide Synthase Inhibitors in Acetaminophen-Induced Hepatic Injury in Mice. Timisoara Med. 2022, 2022(1), 3; doi: 10.35995/tmj20220103.
Received: 12 January 2022 / Accepted: 21 February 2022 / Published: 14 March 2022
Open access
: TIMISOARA MEDICAL JOURNAL is a peer-reviewed open-access journal.Abstract
:(1) Aim of the Study: In this study, we aimed to evaluate the vascular damage and the effects of nitric oxide synthase (NOS) enzyme inhibitors in hepatic damage caused by high doses of acetaminophen (APAP). (2) Material and methods: Fifty-three Swiss albino male mice were used for this study. Hepatic and thoracic aorta toxicity caused by 2 or 6 h exposures to APAP (300 mg/kg intraperitoneally (i.p.)) were evaluated. The general NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME: 25 mg/kg and 50 mg/kg, i.p.) and the neuronal NOS inhibitor 7-nitroindazole (7-NI: 15 mg/kg, i.p.) were administered one hour before APAP exposure. (3) Results: Significant morphological deteriorations were observed after 6 h of APAP exposure in histopathological examinations of hepatic sections. Pre-treatment with L-NAME (at 50 mg/kg) or 7-NI before a 6 h APAP exposure significantly decreased hepatic toxicity (p < 0.05). Significant increases in ALT levels in 6 h of APAP exposure were decreased by both L-NAME (with the 25 mg/kg but not at 50 mg/kg) and 7-NI pre-treatments. No significant change was observed in the measured nitrate/nitrite levels and total antioxidant status in either serum or liver homogenates. No significant deteriorations were observed during either hematoxylin-eosin or immunohistochemical staining in thoracic aorta sections. In the thoracic artery sections, no statistical difference was found in acetylcholine-mediated relaxation, which may indicate endothelial dysfunction. (4) Conclusions: This study demonstrated that APAP-induced hepatic toxicity, especially neuronal NOS inhibitors, may decrease hepatic toxicity. It was also shown that APAP-induced hepatic toxicity was not accompanied by vascular dysfunction.
Keywords:
acetaminophen; hepatic toxicity; nitric oxide synthase inhibitor; 7-nitroindazole1. Introduction
Acetaminophen (APAP) is a commonly prescribed antipyretic and pain-relieving therapeutic. It is known that the side effects of APAP at therapeutic doses (below 4 g daily dose) are low. However, acute high doses and long-term chronic use can cause damage to the liver and lead to fatal outcomes if not treated effectively [1].
APAP is known to be metabolized to the toxic N-acetyl-β-benzoquinone imine (NAPQI), which has high reactivity with cytochrome P450 enzymes in the liver [2,3]. In hepatic cells, NAPQI is effectively detoxified by glutathione [3]. Due to glutathione depletion, at toxic doses of APAP, NAPQI cause necrotic damage in the liver by covalent attachment of centrilobular to intracellular proteins in the liver [4]. In experimental studies carried out on animals, during NAPQI-induced centrilobular liver injury attributed to high-dose APAP exposure, the release of reactive oxygen/nitrogen radicals due to glutathione depletion was observed [5]. The increased reactive oxygen/nitrogen radicals, nitrotyrosine compounds, and inflammatory cytokines cause mitochondrial dysfunction [6,7,8,9]. It has been reported that nitrotyrosine compounds in the liver may be associated with nitric oxide synthesized from sinusoids and the hepatic microcirculation [6]. It has been shown in studies that the activity of inducible nitric oxide synthase (iNOS), which increases nitric oxide levels during APAP toxicity, leads to nitrotyrosine accumulation [6,10,11,12]. However, pharmacological inhibition of iNOS did not decrease APAP-induced liver toxicity in mice [13]. Studies in iNOS null mice have shown a decrease in protein nitration; however, there was also an increase in lipid peroxidation leading to APAP-induced toxicity [14]. A newly developed iNOS inhibitor has been shown to have a reducing effect on APAP-induced hepatic toxicity [15]. Thus, it was thought that iNOS might have a role in protein nitration, but the mechanism of its toxicity is unclear [8]. Delayed hepatic toxicity and the development of fewer nitrotyrosine compounds were shown in wild-type mice with neuronal NOS enzymes compared with untreated wild-type mice, reflecting the role of initial toxicity in APAP-induced hepatic toxicity in mice [16]. Thus, neuronal NOS might be a pathological source of nitric oxide; however, there also might be another independent pathological pathway related to APAP-induced hepatic toxicity [17].
Several authors have proposed that endothelial cell damage and microvascular dysfunction might have a role in the toxicity of APAP [8,11]. Randle and colleagues showed increased catecholamine levels in APAP-induced hepatic toxicity, and decreased toxicity was observed with administration of alpha 1 receptor antagonists, calling attention to the hepatic microcirculation [18]. We aimed to evaluate whether severe macrovascular dysfunction accompanies microvascular dysfunction in APAP-induced hepatic toxicity in mice. Additionally, in this study, we attempted to show the effects of both the non-specific NOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME) and the neuronal NOS inhibitor 7-nitroindazole (7-NI), which may have a role in the hepatic circulation during hepatic injury caused by APAP.
2. Material and Methods
2.1. Animals
The protocol for this study was evaluated by Hacettepe University Faculty of Medicine Experimental Animals Ethics Committee (approval number: 2013-37-05), and this study was carried out in accordance with the “Regulation on the Welfare and Protection of Animals Used for Experimental and Other Scientific Purposes”. In this study, 53 adult, male Swiss albino mice (average weight: 24.1 ± 2.4 g) from the Hacettepe University Medical Faculty Experimental Animal Unit were used. Before the study protocol, mice used in experiments were housed on a 12 h light/dark cycle and with no food or drink restriction.
2.2. Study Protocol
The mice to be used during the experiment were fasted overnight. Subsequently, mice were given intraperitoneal (i.p.) APAP (300 mg/kg, dissolved in warmed saline solution). Measurements were made 2 and 6 h after exposure to APAP. For the control group, the same amount of saline was given. Animals were divided into 3 groups: 2 h of APAP exposure, 6 h of APAP exposure, and a saline control. In order to show the role of the NOS pathway, 1 h prior to administration of APAP, the NOS inhibitor L-NAME (25 mg/kg and 50 mg/kg, i.p.) or the neuronal NOS inhibitor 7-NI (15 mg/kg, i.p.) were given. The mice to be examined were anesthetized with urethane (1.5 g/kg, i.p.) before the experiment.
The hepatic tissues of all animals were photographed before blood samples were taken. The blood was obtained intracardially and then centrifuged (10 min at 1000 g). Obtained samples were stored at −80 °C. Portions of hepatic tissue were prepared fresh and stored at −80 °C until examination. Another piece of hepatic tissue and a piece of the thoracic aorta were stored in 10% formaldehyde solution for histopathological examination. Another part of the thoracic aorta was isolated from the surrounding tissue and kept in physiological saline solution (PSS), which was aerated with a 95% oxygen and 5% carbon dioxide mixture at 37 °C for functional measurements. PSS contained (in mmol/L) NaCl: 118; KCl: 4.6; NaHCO3: 25; MgSO4: 1.2; KH2PO4: 1.2; CaCl2: 1.2; glucose 5.5 and EDTA: 0.025; pH 7.4.
2.3. Pathological Evaluation
The liver and aorta tissues fixed in 10% formaldehyde solution were embedded in paraffin after tissue processing. The paraffin blocks were cut in 5-µm-thick and the sections were stained with haematoxylin-eosin stain. Prepared slides were examined under a light microscope (Olympus, BX46, Tokyo, Japan).
Liver damage was assessed in terms of the following 5 parameters:
- Cytoplasmic vacuolization in hepatocytes;
- Increase in eosinophilia in hepatocytes;
- Hepatocellular necrosis;
- Haemorrhage;
- Periportal area inflammation.
All parameters were scored semiquantitative in 4 categories of severity (scoring scale: 0 = none, 1 = mild, 2 = moderate, 3 = severe).
Thoracic aorta tissue was evaluated by haematoxylin-eosin staining. CD34, a transmembrane glycoprotein synthesized by endothelial cells, has been used to demonstrate damage in vascular endothelial cells. Endothelial cells stained immunohistochemically with a CD34-specific antibody were also examined microscopically.
2.4. Biochemical Measurements
Serum samples were analyzed by using the Alanine Aminotransferase (ALT) Assay Kit (Cayman Chemicals, MI, USA), the Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemicals, MI, USA), and the Total Antioxidant Status Assay Kit (TAS) (Rel Assay, Gaziantep, Turkey). The concentrations of serum ALT, nitrate/nitrite, and TAS are expressed as U/L, µmol/L, and mmol/L, respectively. Liver tissue homogenates were analyzed by using the Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemicals, MI, USA) and the Total Antioxidant Status Assay Kit (Rel Assay, Gaziantep, Turkey). For tissue protein level determination, the spectrophotometric Modified Lowry Protein Assay Kit (Thermo Scientific, MA, CO, USA) were used. The concentrations of hepatic tissue nitrate/nitrite and TAS are expressed as µmol/g protein and mmol/g protein, respectively. All measurements were made under the instruction manual of the kit.
2.5. Functional Measurements
A wire myograph system (610 M Danish Myo-Technology, Aarhus, Denmark) was used, and isometric arterial responses were recorded using a data acquisition system (Powerlab/4SP, AD Instruments, CO, USA). The isolated thoracic aortic rings were cut into lengths of ~2 mm rings. The portion was isolated from the surrounding tissues and was prepared for examination in the myography system. Two steel wires 40 µm in diameter were passed through the lumen of the artery. With the help of the steel wire, the ring was mounted to a 5 mL myograph chamber maintained at 37 °C and continuously aerated with a 95% oxygen and 5% carbon dioxide mixture. The prepared arterial rings were rested for 20 min and then stretched to the calculated normalized diameter step by step using the normalization module program (Danish Myo-Technology, Aarhus, Denmark) as described in the literature [19]. By the end of normalization, the measurements were recorded in mN/mm. After normalization and resting, the arteries were challenged with high K+ by replacing the PSS with 125 mM K+ containing PSS, and the reference contraction curves were recorded. After the contraction reached a plateau, the 125 mM K+ containing PSS was washed out. Then, phenylephrine (1 nM–30 μM) concentration–isometric contraction responses were recorded. The submaximal contraction responses (60–80% of the 125 mM K+ contraction reference value) were recorded. After the submaximal response was obtained, concentration–relaxation response curves of acetylcholine (1 nM–10 μM) added cumulatively to the organ bath were recorded to show endothelium-dependent relaxation responses.
2.6. Reagents and Statistical Analysis
All drugs used in the experiments were obtained from Sigma-Aldrich (St. Louis, MO, USA). The GraphPad Prism (v6, La Jolla, CA, USA) program was used to prepare the graph images and statistics. A non-parametric Kruskal–Wallis test and Dunn’s comparison analysis were used to compare semiquantitative pathological scores. One-way ANOVA for biochemical analyses and Dunnett’s comparative analysis test for multiple group comparisons were used. For repeated measures in aortic responses, two-way ANOVA and Dunnett’s multiple comparison tests were used. p < 0.05 was chosen as the threshold for statistical significance.
3. Results
3.1. Pathological Evaluation
Structural damage was observed macroscopically in livers exposed to APAP for 2 h and 6 h, while no visible damage was observed in the saline-treated control group (Figure 1A–C). Macroscopic improvement of hepatic tissue was observed following administration of 25 mg/kg and 50 mg/kg L-NAME and 7-NI (15 mg/kg), 1 h before APAP (Figure 1D–F).
When liver tissue sections were examined by haematoxylin-eosin staining, histopathologic examination revealed eosinophilia, vacuolisation, hepatocyte necrosis, and diffuse parenchymal haemorrhage in the portal area and near the central vein (Figure 2A–C).
Compared with the control group exposed to APAP for 6 h, when the severity of damage was scored on a 4-point semiquantitative scale, there was a statistically significant impairment in hepatocellular necrosis, haemorrhage, and periportal area inflammation in cytoplasmic vacuolization in hepatocytes (Table 1). Administration of 50 mg/kg L-NAME resulted in a partial improvement similar to that in the control group. Significant improvements were obtained with a 50 mg/kg dose of L-NAME, compared with 6 h of APAP injury. A similar improvement to the control was obtained with pathological assessment results following 7-NI pre-treatment, which was found to be significantly different from 6 h of APAP exposure (Table 1).
Mouse thoracic aortas and endothelial layers were observed as slightly corrupted but not significantly damaged when examined pathologically with haematoxylin-eosin staining. Moreover, there was no significant damage in the endothelial layer with immunohistochemical staining using the endothelial marker CD34. Mild non-significant damage of the endothelial layer seen in the examined sections is shown in Figure 3.
3.2. Biochemical Measurements
As a result of APAP exposure, the measured ALT activity in serum was increased statistically significantly after 6 h of exposure (Figure 4). 7-NI and L-NAME (25 mg/kg) given before APAP resulted in a decrease in measured ALT activity (Figure 4).
No significant changes were observed in the further biochemical parameters measured in both serum and hepatic tissues (Figure 5).
3.3. Functional Evaluation of Thoracic Aorta
Thoracic aortic rings analysed in the myograph system showed no difference in smooth muscle contraction forces (mN/mm) against 125 mM KCl stimulation (Figure 6A). Of the 15 arterial rings (in all 36 arterial rings) were excluded from the study due to the irresponsiveness to phenylephrine concentrations. In the 2 h APAP-induced group, the concentration-contraction curves induced by phenylephrine (1 nM–30 μM) were statistically significantly different (Figure 6B), but there were not significant differences in the relaxation curves obtained by acetylcholine (1 nM–30 μM) in all groups analysed (Figure 6C).
4. Discussion
In this study, hepatic toxicity was observed following systemic administration of high-dose APAP to mice, and it was also shown that pre-administration of the general NOS inhibitor L-NAME and the neuronal NOS inhibitor 7-NI partially reduced hepatic damage and decreased hepatic damage histopathologically, respectively. However, as a result of pharmacological functional and pathologic evaluations, no significant alterations were observed in thoracic aorta.
In this study, hepatic toxicity, which started at 2 h of APAP exposure and became evident at 6 h, was shown in mice macroscopically and with pathological examination using routine haematoxylin-eosin staining. The reported toxicities were thought to be consistent with the literature [3,16]. In the literature, APAP was reported to have a hepatic toxicity beginning and progressing at 4 and 6 h, respectively, after administration of 300 mg/kg gavage or i.p. [11,13]. Additionally, in our study, 300 mg/kg dose of acetaminophen, which causes a moderate level of hepatotoxicity in previous studies, was chosen [13]. Statistically significant deteriorations were observed due to APAP exposure for 6 h in sections of hepatic tissues by using a semiquantitative pathological method. Additionally, 6 h of APAP exposure caused a significant increase in measured ALT levels in mouse serum. The toxicity was attributed to the depletion in glutathione, which led to the uncompromised increase in reactive oxygen/nitrogen species [8]. The complex role of radical nitrogen species in APAP-induced hepatic toxicity was extensively studied, and decreased toxicity to hepatocytes was shown by inhibition of nitric oxide synthase [12]. Additionally, in our study, pre-treatment with L-NAME, the general nitric oxide synthase inhibitor, partially reversed the toxicity caused by APAP in the liver. These improvements in histopathological findings were found to be more prominent in the 50 mg/kg L-NAME than in the 25 mg L-NAME pre-treatment. These results suggest that L-NAME is susceptible to APAP-induced damage reduction. The effect seen at a dose of 25 mg/kg may not have been observed due to shifting the majority toxicity pathway towards superoxide-derived oxidants, as speculated in the literature [13], or due to the significant hemodynamic disturbances that may be caused by L-NAME administered at a dose of 50 mg/kg. In the 25 mg/kg L-NAME and 7-NI pre-treatment groups, mouse serum ALT levels were measured similar to the control group. The partial improvement effect shown for L-NAME has not been shown in the literature [11,13]. In a previous study, NOS inhibitors (N-monomethyl-L-arginine (L-NMMA) (30 mg/kg), L-NAME (2 mg/kg), L-N-(1-iminoethyl)lysine (L-NIL) (3 mg/kg), and aminoguanidine (15 mg/kg)) were not shown to reduce the hepatotoxicity of high doses of APAP in mice [13]. The augmentation of hepatotoxicity by reducing NOS inhibitors appeared to shift the toxicity pathway toward reactive oxygen species [13,14]. However, in our study, L-NAME was administered at high doses of 25 mg/kg and 50 mg/kg, unlike in the literature. It has been thought that, at higher doses, L-NAME might reduce in all other NOS enzymes, which may lead to a complex effect on hepatic toxicity. In addition, the NO releasing and nitrovasodilator properties of L-NAME have been shown in the literature, as well as the exacerbation of these properties during inflammation [20], therefore it is understood that more studies are needed to elucidate the complex effects of L-NAME. In a cell culture study, decreased mitochondrial damage with reduction in the initial phase of cellular damage was evaluated by adding the general NOS inhibitor L-NMMA and the neuronal NOS inhibitor 7-nitroindazole in the re-incubation media, but not with the inducible NOS inhibitor L-NIL (N6-(1-iminoethyl)-L-lysine)) [12]. Therefore, consistent with our results, neuronal NOS might have a prominent role in APAP-induced hepatotoxicity. However, APAP-induced hepatotoxicity was present in NOS2-null mice [16]. Therefore, neuronal NOS is important, but is not the only pathologic pathway in APAP-induced hepatotoxicity. It has also been shown that a newly developed inducible NOS inhibitor had a protective effect against hepatic toxicity caused by APAP [15].
An increase in nitrite/nitrate levels in mouse serums of APAP treatment, which was shown in previous studies [13], could not be shown in our study. We did not observe significant changes between treatment groups. It was thought that if the exposure duration of APAP was increased to 12 or 24 h, significant changes might be observed. This issue might also be related to differences in the animal strains used in our study and the fact that our method lacks the sensitivity to detect differences.
APAP-induced hepatic damage was thought to be related to endothelial cell injury, microvascular dysfunction, and hepatocyte necrosis [6]. In an APAP toxicity model in rats, aortic endothelial- and non-endothelial-induced relaxation responses impaired along with vessel morphology [21]. Therefore, in this study, the inflammation developed by APAP toxicity in mice may also affect the aortic tissue. Here, we tried to show widespread vascular and endothelial impairment. However, in a histopathological examination of vascular tissue with routine haematoxylin-eosin staining, no significant impairment was observed. Besides showing the superficial endothelial cell structure, we assessed CD34 glycoprotein by immunohistochemical staining, but no significant endothelial damage was shown. It was therefore concluded that the 6 h APAP exposure did not cause widespread vascular pathology in a histopathological manner. Although histopathologic damage was not demonstrated in the thoracic aorta, parts of the arterial rings were evaluated in the myograph system to assess the possible damage at the functional receptor level. In our myograph experiments, the phenylephrine-induced contractions were significantly different in the 2 h APAP exposure group. In mice, it was shown that plasma catecholamine levels were increased 5 h after APAP administration, and increased catecholamine levels were associated with APAP-induced hepatotoxicity [18]. Therefore, the hyperactivity of the artery segments in the 2 h APAP group might be related to the early increase in catecholamine concentrations or inflammatory mediators. The increased contractility seen at 2 h of APAP exposure but not at 6 h of APAP exposure may also speculate a down-regulation of post-receptor mechanisms. However, as mentioned before in the results section, few arterial samples were irresponsive to cumulative concentrations of phenylephrine. This irresponsiveness seen in some arteries may be due to the trauma that the tissue was exposed to during preparation for myograph experiments.
5. Conclusions
It was concluded that in APAP-induced hepatic toxicity in mice, a measurable vascular pathology could not be established. In our study, the inability to evaluate the microcirculation in the liver was seen to be a limitation of our study and remains to be established in future studies. In conclusion, this study showed that nNOS in particular may have a clear role in APAP-induced hepatic injury; however, macrovascular functional alterations were not observed. Besides the beneficial effects of nitric oxide synthase inhibitors to be chosen, it is seen that further studies are needed to determine the clinical reflection of the selected doses, as they can change the balance of oxygen radicals and turn the pathological process into a worse situation.
Author Contributions
Conceptualization, E.G.Ç. and N.F.B.; Methodology, E.G.Ç., N.F.B., Ö.İ.Ç, N.Y. and F.B.Ş.; Formal Analysis, E.G.Ç., N.F.B., Ö.İ.Ç, N.Y. and F.B.Ş.; Data Curation, E.G.Ç.; Writing—Original Draft Preparation, E.G.Ç.; Writing—Review and Editing, E.G.Ç., N.F.B., Ö.İ.Ç., F.B.Ş., and G.S.; Supervision, G.S.; Project Administration, E.G.Ç.; Funding Acquisition, E.G.Ç.
Funding
This paper has been granted by the Muğla Sıtkı Koçman University Research Projects Coordination Office through Project Grant Number: 15/065.
Acknowledgments
We would like to thank Lokman Çevik for his technical support in this study.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Bessems, J.; Vermeulen, N.P. Paracetamol (Acetaminophen)-Induced Toxicity: Molecular and Biochemical Mechanisms, Analogues and Protective Approaches. Crit. Rev. Toxicol. 2001, 31, 55–138. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, D.C.; Miwa, G.T.; Lu, A.Y.; Nelson, S.D. N-acetyl-p-benzoquinone imine: A cytochrome P-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. USA 1984, 81, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; Mayeux, P.R.; Hinson, J.A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003, 31, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Jollow, D.J.; Mitchell, J.R.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J. Pharmacol. Exp. Ther. 1973, 187, 195–202. [Google Scholar] [PubMed]
- Cover, C.; Mansouri, A.; Knight, T.R.; Bajt, M.L.; Lemasters, J.J.; Pessayre, D.; Jaeschke, H. Peroxynitrite-Induced Mitochondrial and Endonuclease-Mediated Nuclear DNA Damage in Acetaminophen Hepatotoxicity. J. Pharmacol. Exp. Ther. 2005, 315, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.R.; Kurtz, A.; Bajt, M.L.; Hinson, J.A.; Jaeschke, H. Vascular and Hepatocellular Peroxynitrite Formation during Acetaminophen Toxicity: Role of Mitochondrial Oxidant Stress. Toxicol. Sci. 2001, 62, 212–220. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; Lamps, L.W.; McCullough, S.; A Hinson, J. Interleukin 6 and hepatocyte regeneration in acetaminophen toxicity in the mouse. Biochem. Biophys. Res. Commun. 2003, 309, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of Acetaminophen-Induced Liver Necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405. [Google Scholar] [CrossRef]
- Hou, H.-S.; Liao, C.-L.; Sytwu, H.-K.; Liao, N.-S.; Huang, T.-Y.; Hsieh, T.-Y.; Chu, H.-C. Deficiency of Interleukin-15 Enhances Susceptibility to Acetaminophen-Induced Liver Injury in Mice. PLoS ONE 2012, 7, e44880. [Google Scholar] [CrossRef]
- Gardner, C.R.; Heck, D.E.; Yang, C.S.; Thomas, P.E.; Zhang, X.-J.; DeGeorge, G.L.; Laskin, J.D.; Laskin, D.L. Role of nitric oxide in acetaminophen-induced hepatotoxicity in the rat. Hepatology 1998, 27, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Abril, E.R.; Bethea, N.W.; McCuskey, R.S. Role of nitric oxide in hepatic microvascular injury elicited by acetaminophen in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G60–G67. [Google Scholar] [CrossRef]
- Burke, A.S.; MacMillan-Crow, L.A.; Hinson, J.A. Reactive Nitrogen Species in Acetaminophen-Induced Mitochondrial Damage and Toxicity in Mouse Hepatocytes. Chem. Res. Toxicol. 2010, 23, 1286–1292. [Google Scholar] [CrossRef]
- Hinson, J.A.; Bucci, T.J.; Irwin, L.K.; Michael, S.L.; Mayeux, P.R. Effect of Inhibitors of Nitric Oxide Synthase on Acetaminophen-Induced Hepatotoxicity in Mice. Nitric Oxide 2002, 6, 160–167. [Google Scholar] [CrossRef]
- Michael, S.L.; Mayeux, P.R.; Bucci, T.J.; Warbritton, A.R.; Irwin, L.K.; Pumford, N.R.; Hinson, J.A. Acetaminophen-Induced Hepatotoxicity in Mice Lacking Inducible Nitric Oxide Synthase Activity. Nitric Oxide 2001, 5, 432–441. [Google Scholar] [CrossRef]
- Kamanaka, Y.; Kawabata, A.; Matsuya, H.; Taga, C.; Sekiguchi, F.; Kawao, N. Effect of a potent iNOS inhibitor (ONO-1714) on acetaminophen-induced hepatotoxicity in the rat. Life Sci. 2003, 74, 793–802. [Google Scholar] [CrossRef]
- Agarwal, R.; MacMillan-Crow, L.A.; Rafferty, T.M.; Saba, H.; Roberts, D.W.; Fifer, E.K.; James, L.P.; Hinson, J.A. Acetaminophen-Induced Hepatotoxicity in Mice Occurs with Inhibition of Activity and Nitration of Mitochondrial Manganese Superoxide Dismutase. J. Pharmacol. Exp. Ther. 2010, 337, 110–118. [Google Scholar] [CrossRef]
- Agarwal, R.; Hennings, L.; Rafferty, T.M.; Letzig, L.G.; McCullough, S.; James, L.P.; MacMillan-Crow, L.A.; Hinson, J.A. Acetaminophen-Induced Hepatotoxicity and Protein Nitration in Neuronal Nitric-Oxide Synthase Knockout Mice. J. Pharmacol. Exp. Ther. 2011, 340, 134–142. [Google Scholar] [CrossRef]
- Randle, L.; Sathish, J.; Kitteringham, N.R.; Macdonald, I.; Williams, D.P.; Park, B.K. α1 -Adrenoceptor antagonists prevent paracetamol-induced hepatotoxicity in mice. J. Cereb. Blood Flow Metab. 2008, 153, 820–830. [Google Scholar] [CrossRef]
- Mulvany, M.J.; Halpern, W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977, 41, 19–26. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, M.; Mukosera, G.T.; Borchardt, D.; Li, Q.; Tipple, T.; Ahmed, A.; Power, G.G.; Blood, A.B. L-NAME releases nitric oxide and potentiates subsequent nitroglycerin-mediated vasodilation. Redox Biol. 2019, 26, 101238. [Google Scholar] [CrossRef]
- Porto, H.K.P.; Grando, M.D.; Ramalho, L.N.Z.; Valadares, M.C.; Bendhack, L.M.; Batista, A.C.; Rocha, M.L. Exposure to acetaminophen impairs vasodilation, increases oxidative stress and changes arterial morphology of rats. Arch. Toxicol. 2019, 93, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Photographic images of the liver. Top line: (A) Saline-treated control mice; (B): 2 h of APAP exposure; (C): 6 h of APAP exposure; Bottom line: (D): 25 mg/kg L-NAME *; (E): 50 mg/kg L-NAME *; and (F): 15 mg/kg 7-NI * (* 1 h before 6 h APAP exposure i.p.).
Figure 1.
Photographic images of the liver. Top line: (A) Saline-treated control mice; (B): 2 h of APAP exposure; (C): 6 h of APAP exposure; Bottom line: (D): 25 mg/kg L-NAME *; (E): 50 mg/kg L-NAME *; and (F): 15 mg/kg 7-NI * (* 1 h before 6 h APAP exposure i.p.).
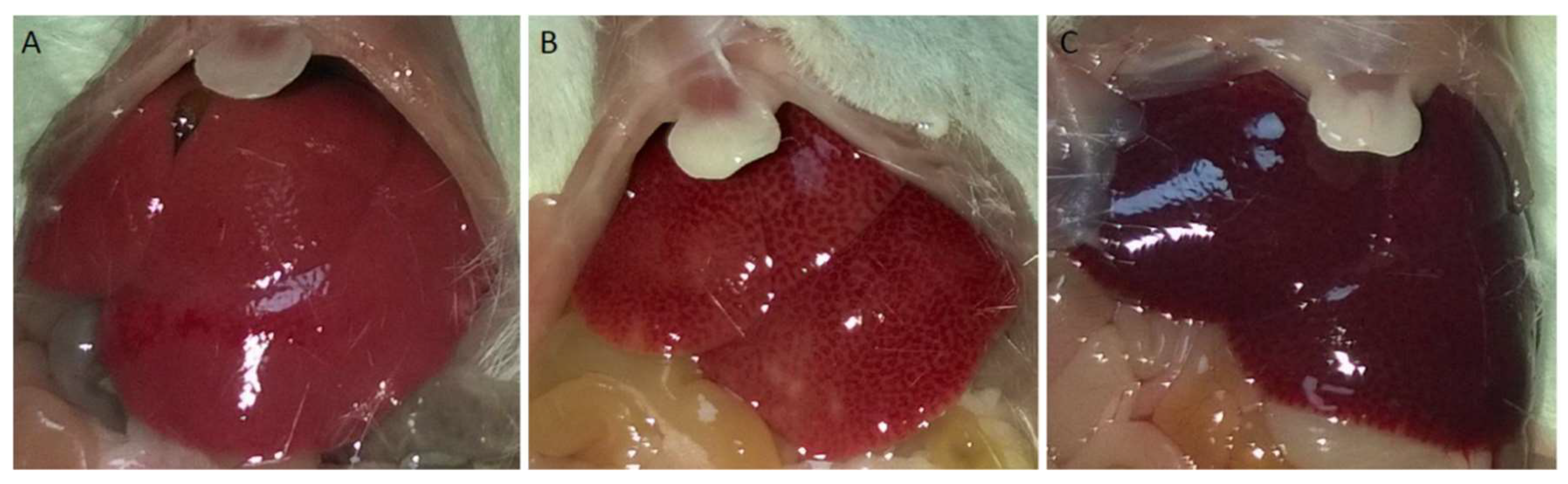
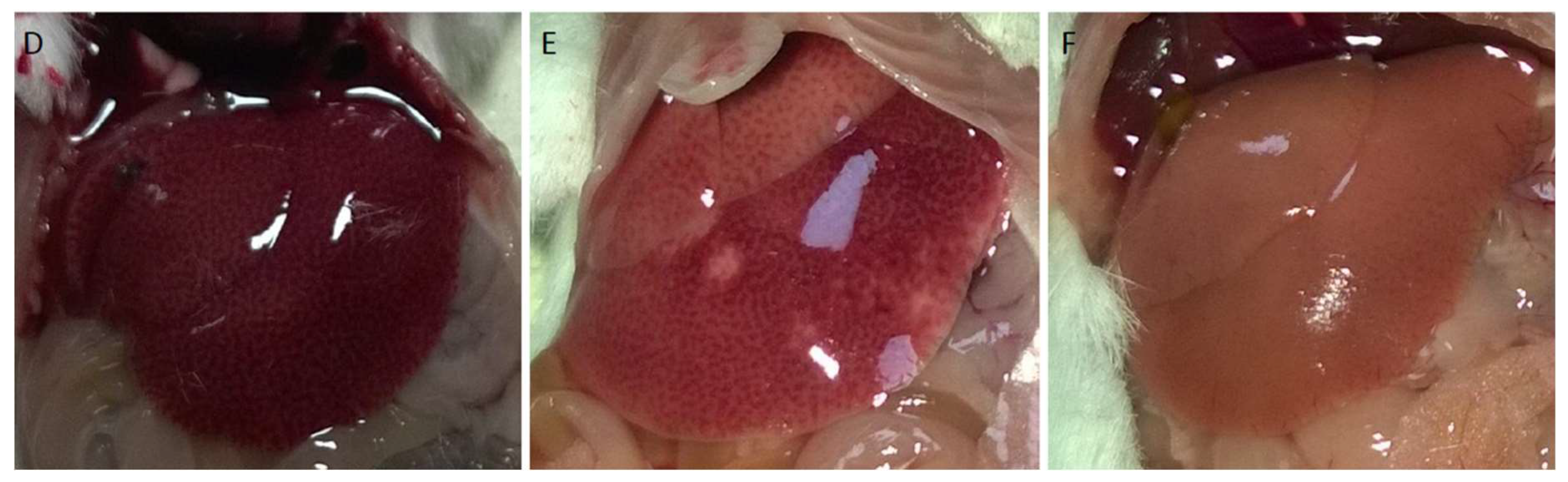
Figure 2.
Representative microscopic examination images of hepatic tissue. (A) Control liver tissue, normal central vein (large arrow) and hepatocytes (small arrow). Surrounding sinusoidal arrangement and portal area (in circle) without inflammation are observed. (B) Inflammation (stars) that from one portal area to another and towards the central venules. (C): Dysregulation of hepatocytes around the central vein, cytoplasmic vacuolization (arrows), necrosis of hepatocytes (stars), and diffuse parenchymal haemorrhage (triangles). Haematoxylin-eosin staining, x100.
Figure 2.
Representative microscopic examination images of hepatic tissue. (A) Control liver tissue, normal central vein (large arrow) and hepatocytes (small arrow). Surrounding sinusoidal arrangement and portal area (in circle) without inflammation are observed. (B) Inflammation (stars) that from one portal area to another and towards the central venules. (C): Dysregulation of hepatocytes around the central vein, cytoplasmic vacuolization (arrows), necrosis of hepatocytes (stars), and diffuse parenchymal haemorrhage (triangles). Haematoxylin-eosin staining, x100.
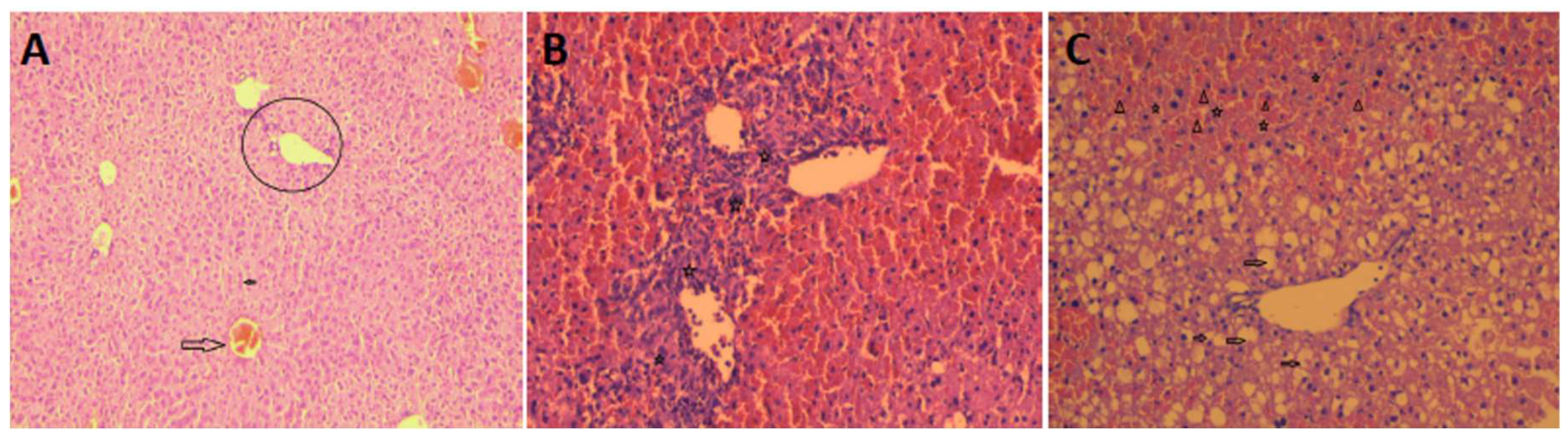
Figure 3.
Microscopic findings of mouse thoracic aorta sections. (A) Normal vascular structure with intact endothelial cells (arrows) (haematoxylin-eosin, 100×). (B) Mild severe damage and spillage (arrows) in endothelial cells (haematoxylin-eosin, 200×). (C) Demonstration of endothelial cells immune-histochemically using the marker CD34. Normal endothelial cells (large arrows) and areas that are not stained due to spilled endothelial cells (small arrows) (CD34, 200×) describes mild damage and spillage in endothelial cells.
Figure 3.
Microscopic findings of mouse thoracic aorta sections. (A) Normal vascular structure with intact endothelial cells (arrows) (haematoxylin-eosin, 100×). (B) Mild severe damage and spillage (arrows) in endothelial cells (haematoxylin-eosin, 200×). (C) Demonstration of endothelial cells immune-histochemically using the marker CD34. Normal endothelial cells (large arrows) and areas that are not stained due to spilled endothelial cells (small arrows) (CD34, 200×) describes mild damage and spillage in endothelial cells.
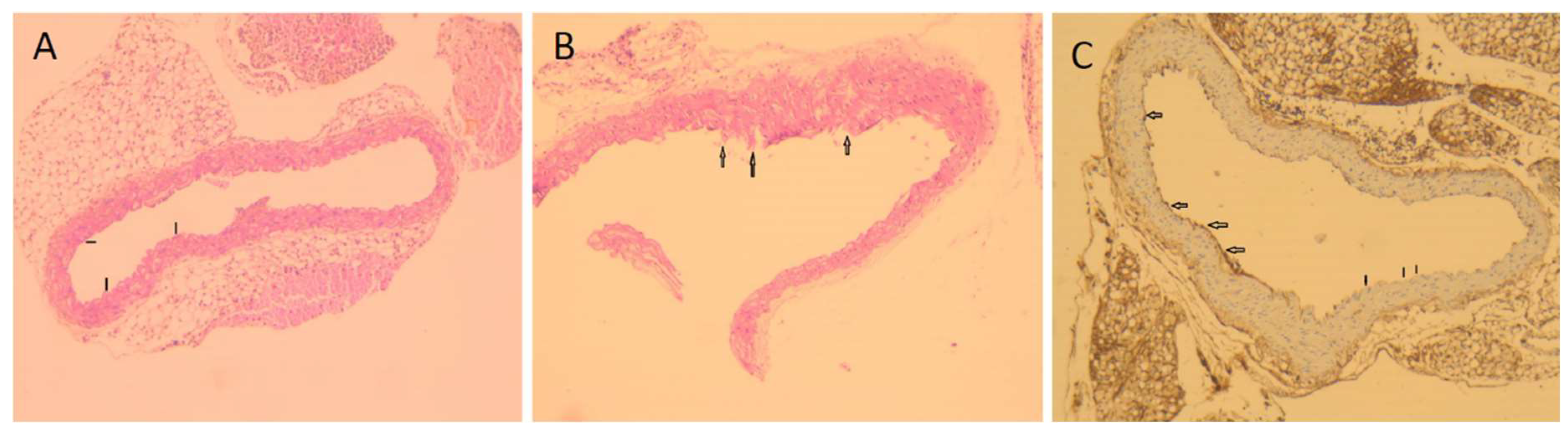
Figure 4.
L-NAME 25 mg/kg (n = 6) 1 h before 6 h APAP exposure, 6 h APAP exposure (n = 6), 2 h APAP exposure; L-NAME 50 mg/kg (n = 5) 1 h prior to 6 h of APAP exposure; 7-NI 15 mg/kg (n = 4) 1 h prior to 6 h of APAP exposure. *: p < 0.05 (when compared with the control).
Figure 4.
L-NAME 25 mg/kg (n = 6) 1 h before 6 h APAP exposure, 6 h APAP exposure (n = 6), 2 h APAP exposure; L-NAME 50 mg/kg (n = 5) 1 h prior to 6 h of APAP exposure; 7-NI 15 mg/kg (n = 4) 1 h prior to 6 h of APAP exposure. *: p < 0.05 (when compared with the control).
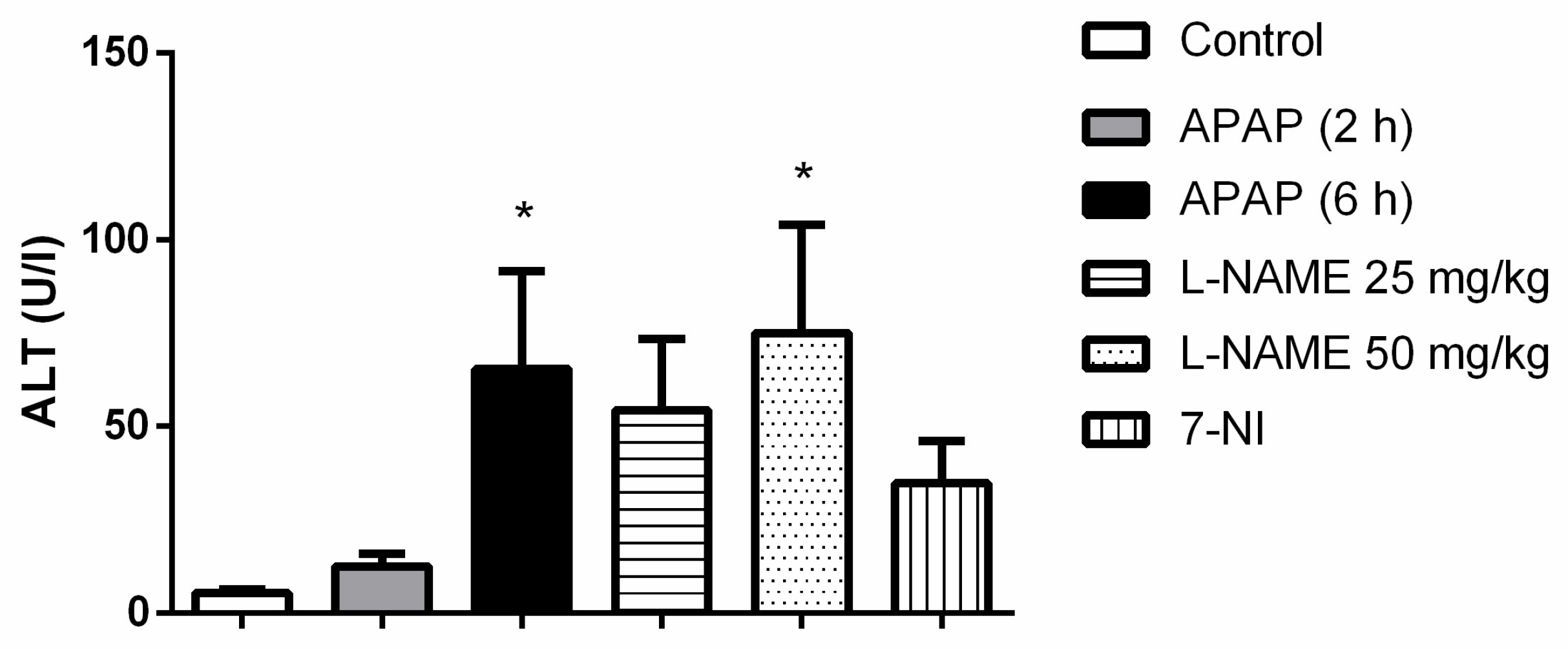
Figure 5.
Biochemical measurements in serum samples and liver tissues. Top row: nitrite/nitrate (A) and total antioxidant level (B) measured in mouse sera. Control: Saline control group (n = 7). APAP (2 h): 2 h exposure of APAP (n = 10). APAP (6 h): 6 h exposure of APAP (n = 6). Lower row: nitrite/nitrate level (C) and total antioxidant level (D) measured in mouse liver tissue. Control: Saline control group (n = 9). APAP (2 h): 2 h exposure of APAP (n = 14). APAP (6 h): 6 h exposure of APAP (n = 13).
Figure 5.
Biochemical measurements in serum samples and liver tissues. Top row: nitrite/nitrate (A) and total antioxidant level (B) measured in mouse sera. Control: Saline control group (n = 7). APAP (2 h): 2 h exposure of APAP (n = 10). APAP (6 h): 6 h exposure of APAP (n = 6). Lower row: nitrite/nitrate level (C) and total antioxidant level (D) measured in mouse liver tissue. Control: Saline control group (n = 9). APAP (2 h): 2 h exposure of APAP (n = 14). APAP (6 h): 6 h exposure of APAP (n = 13).
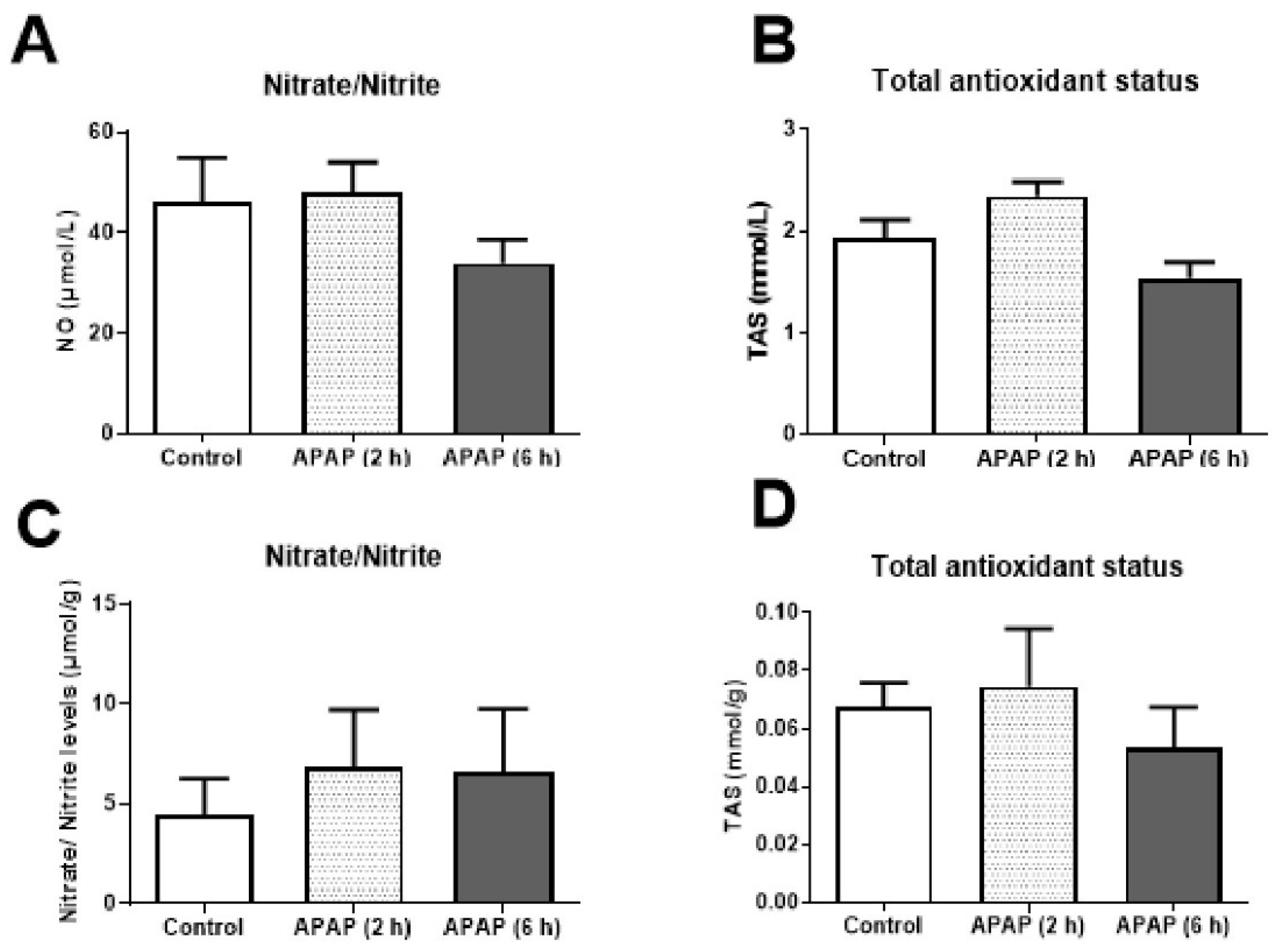
Figure 6.
Functional experiments in the thoracic aorta. (A) Contractions of the thoracic aorta for 125 mM KCl stimulation. (B) The concentration–contraction curve induced by phenylephrine (1 nM–30 μM). (C): The relaxation curve induced by acetylcholine (1 nM–30 μM) in submaximally contracted tissues. 2 h APAP (n = 6), 6 h APAP (n = 4), Control group (n = 5). * p < 0.05 was considered statistically significant when compared with the control group.
Figure 6.
Functional experiments in the thoracic aorta. (A) Contractions of the thoracic aorta for 125 mM KCl stimulation. (B) The concentration–contraction curve induced by phenylephrine (1 nM–30 μM). (C): The relaxation curve induced by acetylcholine (1 nM–30 μM) in submaximally contracted tissues. 2 h APAP (n = 6), 6 h APAP (n = 4), Control group (n = 5). * p < 0.05 was considered statistically significant when compared with the control group.
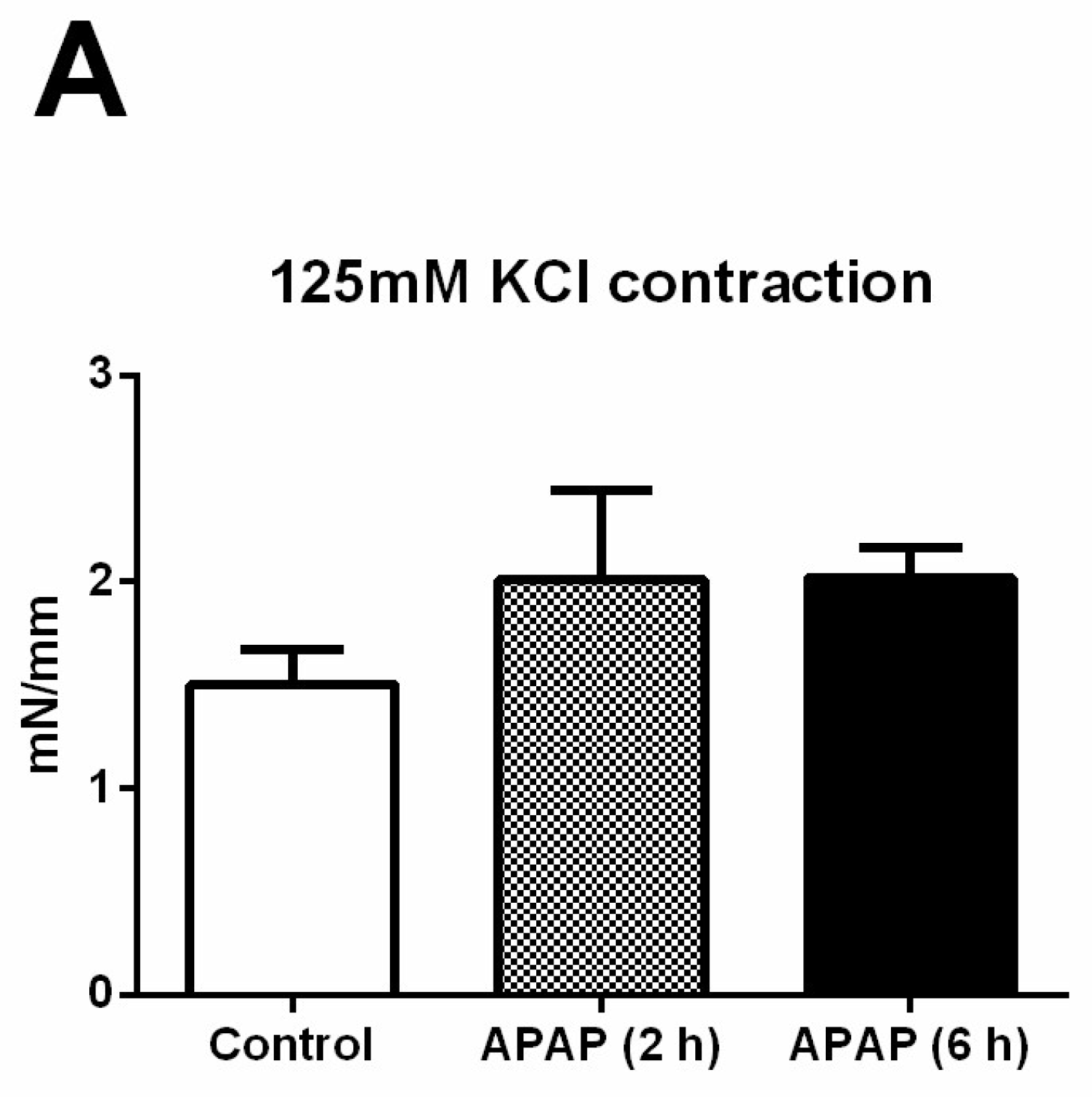
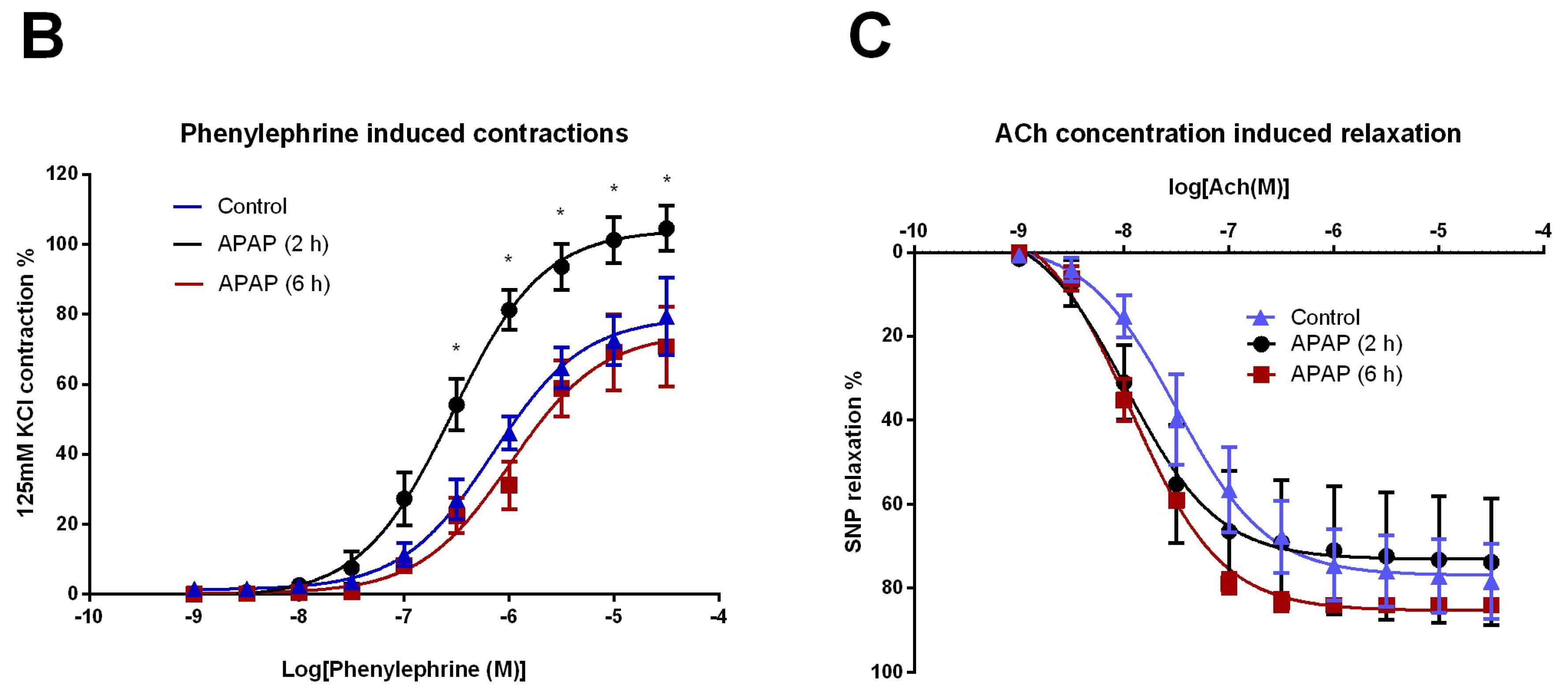

Table 1.
Pathological evaluation of liver.
| Control n = 5 | APAP (2 h) n = 8 | APAP (6 h) n = 8 | L-NAME 25 mg/kg n = 5 | L-NAME 50 mg/kg n = 4 | 7-NI 15 mg/kg n = 5 | |
|---|---|---|---|---|---|---|
| Cytoplasmic vacuolization in centrilobulary hepatocytes | 1 (0–1) | 1 (0.75–2.25) | 2.25 (2–3) * | 2 (1–2) | 2 (1–3) | 1 (0–1) † |
| Increase in eosinophilia in centrilobular hepatocytes | 0 (0–0) | 0 (0–0) | 0 (0–0.25) | 0 (0–0) | 0 (0–0) | 0 (0–0) |
| Hepatocellular necrosis | 0 (0–0) | 0 (0–1,25) | 2 (1–2) * | 2 (1–2) * | 1 (0–2,25) | 0 (0–0) † |
| Haemorrhage | 0 (0–0) | 0.5 (0–1.25) | 3 (3–3) * | 3 (1–3) * | 1.5 (0–3) | 0 (0–0) † |
| Periportal area inflammation | 0 (0–0) | 0 (0–0.25) | 1.5 (1–2.25) * | 1 (0–1) | 0.5 (0–1.25) | 0 (0–0) † |
Values were given as median (25–75% percentile). Assessment was made by scoring 0: none 1: mild 2: moderate 3: severe. * p <0.05 (when compared with control group); † p <0.05 (when compared to the group with 6 h acetaminophen exposure).
© 2022 Copyright by the authors. Licensed as an open access article using a CC BY 4.0 license.